
.png
)
{kind=link}
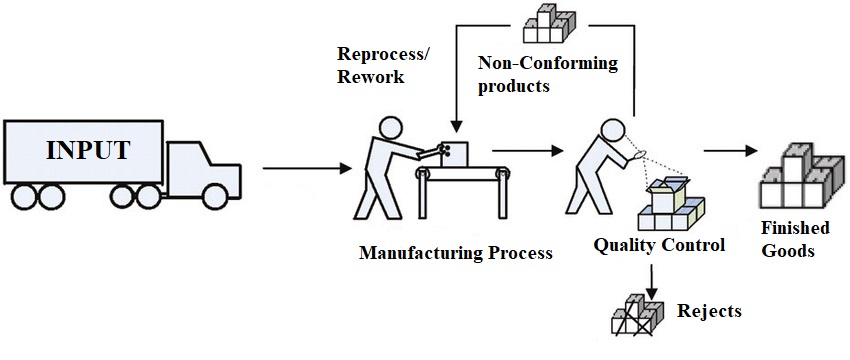
There is a popular phrase that says “Mistakes are bound to happen”, so do the non-conformances in the pharmaceutical manufacturing. Even if you have validated processes, qualified vendors, qualified equipment, calibrated instruments, in-process checks, SOPs etc. in place, possibility of observing non-conformance or failure to meet pre-defined specifications may not be ruled out completely.
So, does it mean that the entire batch has to be destroyed? What if the product is too costly that a manufacturing firm is not ready to bear the losses? What if some activities like reprocessing or reworking, performed in the ambit of regulations may save the batch? Can we perform reprocessing or reworking on every batch? What is the difference between reprocessing and reworking? If reprocessing or reworking is applicable to manufacturing of APIs only or can it be used in case of finished formulation also?
Let’s start answering all the concerns that come to the mind of any manufacturer, beginning with the difference between reprocessing and reworking.
Reprocessing: The intermediate or a batch of a product (Part or full) which upon failing to meet the predefined specification may be introduced again to a previous step in the validated manufacturing process. This process of reintroducing the non-conforming material into the previous step of a validated manufacturing step to get a product meeting all the predefined specifications is called Reprocessing. Repeating a filtration or a crystallization step or other appropriate chemical or physical manipulation steps (e.g., distillation, chromatography, milling) that are part of the established manufacturing process to improve the product characteristics or to make it meet the predefined specifications are considered acceptable.
Reprocessing procedures are foreseen as occasionally necessary for biological medicines and, in such cases, are validated and pre-approved as part of the marketing authorization.
Reworking: The intermediate or a batch of a product (Part or full) which upon failing to meet the predefined specification may be subjected to a different manufacturing process which is not the part of previously validated manufacturing process. This process of subjecting the non-conforming material to a different manufacturing process to get a product meeting all the predefined specifications is called Reworking. Adding additional purification or milling steps etc. that are not the part of the established manufacturing process to improve the product characteristics or to make it meet the predefined specifications are categorized as reworking.
As the reworking arises due to unexpected outcome, hence, the same cannot be pre-approved as part of the marketing authorization.
Points to remember (Reprocessing):
- Upon observing any non-conformance, it should be immediately brought to the knowledge of Quality Unit and shall be escalated to the management at the higher level.
- An investigation may be carried out to know the exact reason for the failure.
- Evaluating the type and extent of non-conformance and considering the outcome of the investigation, the quality unit after discussion with cross functional team and management may approve or reject the proposal of reprocessing.
- Permission for carrying out reprocessing shall be granted on following conditions:
- Reprocessing procedure shall be approved by Quality Unit beforehand.
- The quality of the finished products shall not be affected.
- New batch number shall be allotted to the reprocessed batch. If possible distinct batch number shall be given for easy identification.
- If required Quality control may perform additional testing to ascertain the quality of the product.
- The firm shall carry out proper risk assessment before initiating the reprocessing. Potential formation of by-products and over reacted materials shall be considered.
- The entire reprocessing procedure shall be thoroughly documented.
- Wherever required the reprocessing steps may be validated.
- Reprocessing shall be considered as a rare event.
- It should be noted that in event of repeated reprocessing of similar nature for majority of batches of a product, then the reprocessing may be included as part of the batch manufacturing process.
- The reprocessed batch shall be thoroughly evaluated on all the quality parameters before its release.
- Normally reprocessed batches do not require regulatory approval before product release, but the same shall be included in the Annual notifications to the regulatory authority.
Points to remember (Reworking):
- Reworking shall be considered only in exceptional circumstances and shall be avoided as much as possible.
- Upon observing any non-conformance, it should be immediately brought to the knowledge of Quality Unit and shall be escalated to the management at the highest level.
- An investigation may be carried out to know the exact reason for the failure.
- Evaluating the type and extent of non-conformance and considering the outcome of the investigation, the quality unit after discussion with cross functional team and management may approve or reject the proposal of Reworking.
- Permission for carrying out reworking shall be granted on following conditions:
- Reworking procedure shall be approved by Quality Unit beforehand.
- The quality of the finished products shall not be affected. Documentation shall be done to show that the reworked product is of equivalent quality to that produced by the original process.
- New batch number shall be allotted to the reworked batch. If possible distinct batch number shall be given for easy identification.
- If required Quality control may perform additional testing to ascertain the quality of the product. Where routine analytical methods are inadequate to characterize the reworked batch, additional methods should be used.
- The firm shall carry out proper risk assessment before initiating the reworking.
- The entire reworking procedure shall be thoroughly documented.
- The firm shall carryout concurrent validation. The protocol shall define the rework procedure, how it will be carried out, and the expected results. If there is only one batch to be reworked, a report can be written and the batch released once it is found to be acceptable.
- The reworked batch shall be thoroughly evaluated on all the quality parameters before its release.
- Reworked batch shall be placed on stability testing as per ongoing stability program.
- Procedures should provide for comparing the impurity profile of each reworked batch against batches manufactured by the established process.
- Normally reworked batches shall be released after regulatory approval for the variation.
Reworking in case of Finished formulations: It is learnt that most of the regulatory authorities are not so keen in carrying out any reworking in case of finished formulations, however, they are pretty comfortable for Reprocessing. Reprocessing may be allowed provided that it is done in exceptional circumstances, after proper investigation, the finished product is of comparable quality to the original product etc. for example carrying out sifting again or may be repacking after deblistering.
In few cases recovery is also allowed. Recovery is defined as the introduction of all or part of previous batches (or of re-distilled solvents and similar products) of the required quality into another batch at a defined stage of manufacture. It includes the removal of impurities from waste to obtain a pure substance or the recovery of used materials for a separate use. The recovery should be carried out in accordance with a defined procedure after evaluation of the risks involved, including any possible effect on shelf-life. The recovery should be recorded.
Important Considerations:
- USFDA and EU does allow reworking in case of API but for finished formulations reworking is not stated in their respective guidelines.
- Introducing unreacted material back into a process and repeating a chemical reaction is considered to be reprocessing unless it is part of the established process. Such reprocessing should be preceded by careful evaluation to ensure that the quality of the intermediate or API is not adversely impacted due to the potential formation of by-products and over-reacted materials.
- Continuation of a process step after an in-process control test has shown that the step is incomplete is considered to be part of the normal process. This is not considered to be reprocessing.
- Materials to be reprocessed or reworked should be appropriately controlled to prevent unauthorized use
- It should be noted that special care shall be taken for the products which are returned from the market. These requires special attention as they were out of manufacturer control after their dispatch from manufacturing premises. They should be destroyed unless without doubt their quality is found satisfactory. They may be considered for re-sale, re-labelling or recovery in a subsequent batch only after they have been critically assessed by the Quality Control Department in accordance with a written procedure. The nature of the product, any special storage conditions it requires, its condition and history, and the time elapsed since it was issued should all be taken into account in this assessment. Where any doubt arises over the quality of the product, it should not be considered suitable for re-issue or re-use, although basic chemical reprocessing to recover active ingredient may be possible. Any action taken should be appropriately recorded.
References:
- Section 211.115 of 21 CFR of USFDA
- Guidance for Industry, Q7A Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients
- WHO Technical Report Series no. 986, Annexure 2
- EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use; Volume 4, Part I, Chapter 5, Production
- EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use; Volume 4, Part II, Basic Requirements for Active Substances used as Starting Materials
- ICH Q7, Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
- Schedule M of Drugs and Cosmetics Act, 1940